Here you are going to find short summaries of selected projects that I led or was involved in. If you are interested in more information don't hesitate to reach out to me.
Project Description
In early 2022 I embarked on a start-up project at the University of Heidelberg with my former PhD supervisor Prof. Michael Knop and two of his team members, Dr. Hans Hombauer, previously PI at DKFZ, and Dr. Dan Lou, previously at the CDC in Shanghai, China, and Johns Hopkins University, USA. We got in-depth assistance by Daniel Gau, consultant and life science company builder and received bits of help and advice by a lot of other people.
The idea: We developed an antibody detection platform called "Seromux" with both antibody and sample multiplexing capabilities. Antibody multiplexing means that we could detect and quantify potentially hundreds of different antibodies in one sample. Sample multiplexing means that many samples could be processed in one detection step rather than relying on one separate detection step for each sample separately. The two main ingredients of Seromux were the yeast display techology and FACS. However, we substantially advanced the yeast display technology drawing on advanced high-throughput genetic and strain engineering techniques previously developed in the Knop group, resulting in producing and optimizing the expression properties of hundreds of yeast strains in parallel. Demultiplexing and quantification of the signal was based on barcodes and deep sequencing (REF to Ilia's paper). Such an approach enables determining complex antibody profiles for hundreds of people in short time frames enabling to answer novel questions in fields as diverse as autoimmunity, cancer immunology and cancer therapy, personalized medicine and infectious disease and vaccine research.
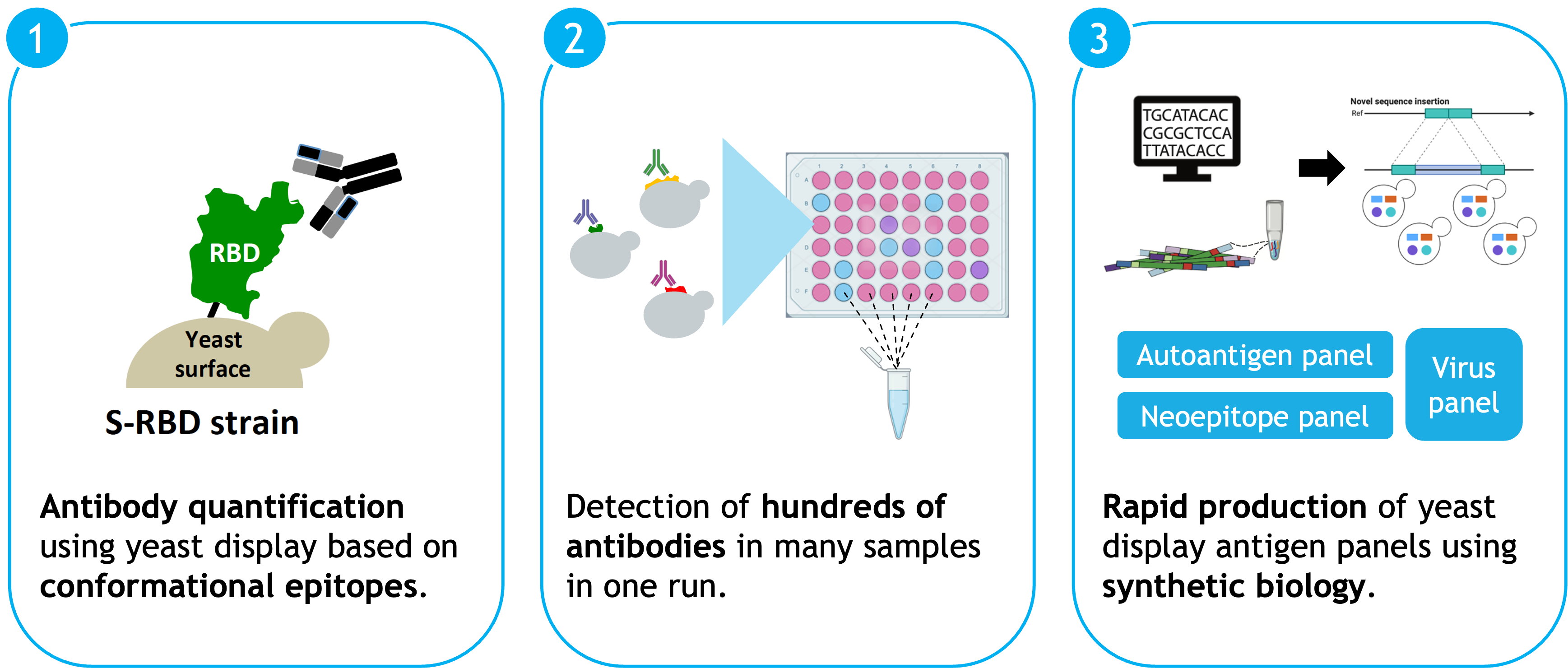
Outcome and Relevance
We successfully applied the Seromux technology to perform multiplexed detection of SARS-CoV-2-specific antibodies in a 96-well plate format. Encouraged by our results we applied for the prestigious "EXIST Forschungstransfer" funding by the Federal Ministry for Economic Affaris and Climate Action of Germany and made it onto the shortlist of the EXIST jury. While the jury was convinced by the technical aspects and our team, they unfortunately considered it still too early for EXIST's more late-stage funding scope. Eventually we suffered from the "hen and egg" problem that we needed more money to provide more convincing data concerning the feasibility while investors and funding body expected such data in order to be convinced of an investment in the first place. Therefore, the project was unfortunately terminated due to a lack of financing.
Contact
If you are interested to learn more about this project, get in touch!
Project Description
This was one of my side projects but I have found it very fascinating and instructive.
Have you ever wondered how a computer works? That is, from the ground up: how do electrical 0 and 1 signals end up executing complex computer programs? This book provides an answer: starting with a hardware simulator and nothing more than a "not and" ("nand") gate the reader goes through all of the steps involved in building a modern computer and eventually implements an object-oriented programming language and an operating system. This is done by guiding the reader through twelve chapters, each containing one project. Starting with a hardware simulator one first designs all other logic gates from the nand gate, then learns how to do arithmetic and logical operations with those gates and then builds a CPU with data and instruction memory access. After that the journey continues: an assembly language is specified and implemented and an assembler is programmed. The next project is the implementation of a virtual machine which is conceptually modelled after the Java virtual machine. This virtual machine serves as the backend of a compiler that the reader implements for the programming language "Jack" (which is specific to the book). The last project is the implementation of a simple operating system.
A fascinating book giving you an idea of the "thousand layers of abstraction" that our interactions with computers require and helping to understand how these wonderful machines work. At the same time, the challenging projects train one's ability to "abstract away" technical details layer by layer.
Project Description
In this project at Bayer I used self-supervised learning with convolutional neural networks on cell painting data with the aim to extract embeddings that are informative of drug mode of action. This project was part of the JUMP-Cell Painting consortium coordinated by the Broad Institute and comprising several partners from academia and the pharmaceutical industry.
Technologies and Methods
- Deep learning (self-supervised learning) using convolutional neural networks and derivation of embeddings.
- PyTorch and the SciPy stack.
- Image processing.
Links to Resources
Project Description
In this project at Bayer I used machine learning in combination with chemoinformatics tools to predict compound activity. Another important aspect was the usage of SHAP values to map model explanations to certain substructures. The latter was integrated into an open soruce tool called "CIME" (ChemInformatics Model Explorer) which enables visualizing "which areas of the compounds have the greatest influence on the outcome" (Humer et al., 2021).
Technologies and Methods
- Various machine learning methods for cheminformatics.
- Scikit learn and the SciPy stack.
- Cheminformatics using the RDKit Python package.
Links to Resources
Project Description
Antibiotic resistance has become a major public health threat and is increasing at a rapid rate. One reason for the large number of failures in clinical trials is ignorance of drug mode of action (MoA). Many studies have tried to infer drug MoA from experimental data and have provided valuable insights. However, they are either based on only a handful of drugs or require laborious/expensive (omics-)methods.
In this project we aimed to use chemical genetics data of 71 different antibacterial compounds measured at different concentrations to predict drug MoA in Escherichia coli. The underlying data set was taken from chemical genetics experiments of Nichols and colleagues (2011) and additional experimental data collected in the Typas lab. In chemical genetics, a knockout collection of non-essential E. coli genes is subjected to a sub minimal inhibitory concentration (MIC) drug dosage to identify mutants that are more sensitive or more resistant to the tested drug. This is known as negative and positive drug-gene interaction, respectively. Doing this at a genome-wide scale results in a high-dimensional "phenotypic" fingerprint for each drug that can inform on drug MoA.
We used this data set to train a machine learning algorithm using the physiological process disrupted by the drug as the target variable. Importantly, our aim was not only to achieve great accuracy but also to identify a selected number of representative "MoA fingerprint" genes that are sufficient to predict drug MoA. This was evaluated on a separate test set. The idea behind this was that algorithmic prediction of MoA is only beneficial in drug screening procedures if the necessary experimental data are quick and easy to collect.
In the second part of the project we performed follow-up experiments to validate selected model predictions. Most of our efforts were focused on understanding the MoA of thiolutin, a compound that our model predicted to interfere with bacterial cell wall biosynthesis. This is in contrast to the conventional belief that thiolutin acts as an RNA polymerase inhibitor. However, recent research suggests that thiolutin's primary MoA may be a result of chelating zinc (e.g. Lauinger et al., 2017). Using assays to monitor growth and cell lysis under different conditions and genetic backgrounds as well as thermal proteome profiling (TPP) we tested if there is a connection between zinc and other cations and the bacterial cell wall machinery that might be disrupted by thiolutin.
Technologies and Methods
- Setup of a machine learning pipeline with nested cross-validation and different performance metrics and loss functions.
- Gradient boosted trees, elastic net, lasso, ridge regression, neural networks.
- R, Python, Git, Databases.
- Feature extraction, feature engineering, feature selection.
- Clustering and dimensionality reduction techniques.
- Cheminformatics (physicochemical properties and fingerprints).
- Quantitative image processing and object tracking in microfluidics chambers.
- Analysis and interpretation of bacterial growth curves.
- Interpretation of chemical genetics data sets and drug-gene interactions.
- Genetic analysis of mutant phenotypes in relation to drug mode of action.
- Thermal proteome profiling (see for example this paper).
Links to Resources
- Link to the GitHub repository
- Coming soon: link to the manuscript (waiting for review by PI).
Credits
The project received substantial help and input by Stefan Bassler and Athanasios Typas as well as Georg Zeller and Léonard Dubois. Additional support was given by Michael Knopp, André Mateus, Mikhail M. Savitski, and KC Huang.
Project Description
The advent of high-throughput microarray and DNA/RNA sequencing technologies revealed the presence of many previously unknown classes of non-coding RNAs. In the yeast Saccharomyces cerevisiae one class of these transcripts were termed "stable unannotated transcripts" (SUTs), many of which overlap protein-coding genes in antisense direction. In this project we tried to understand the global impact of these antisense SUTs on protein abundance by seamlessly inserting unidirectional transcriptional terminators after GFP-tagged genes in 188 separate yeast strains. Subsequently, protein levels were measured using high-throughput quantitative microscopy and colony fluorescence measurements. This helped answer questions concerning the impact of antisense SUTs on protein levels, revealed factors predictive of antisense functionality and revealed a previously unknown mechanism of antisense-dependent gene regulation.
In addition to our scientific findings the project also resulted in a gene duplication method in yeast (see references below).
Technologies and Methods
- High-throughput quantitative microscopy at high accuracy (over 500 strains in 4 different growth conditions), image processing, and corresponding statistical analyses.
- High-throughput genetics and strain engineering.
- R, Python, ImageJ, Bash, data analysis pipelines.
- High-throughput FACS and analysis.
- Genetic analysis of regulatory genetic elements.
- Genomics (tiling arrays and ChIP-Seq) and other omics data integration.
- Nucleic acid biochemistry such as RT-qPCR, Northern blotting (including labelling pulse-chase experiments), 3'-RACE. Quantitative immunoblotting.
Links to Selected Resources
You can find all papers under my ORCID iD. The following are the (shared) first author publications from my PhD:
- Huber, F.*, Bunina, D.*, Gupta, I., Khmelinskii, A., Meurer, M., Theer, P., Steinmetz, Lars M., Knop, M. Protein Abundance Control by Non-coding Antisense Transcription. Cell Reports 2625-2636 (2016).
- Bunina, D.*, Stefl, M.*, Huber, F.*, Khmelinskii, A., Meurer, M., Barry, J.D., Kats, I., Kirrmaier, D., Huber, W., and Knop, M. Upregulation of SPS100 gene expression by an antisense RNA via a switch of mRNA isoforms with different stabilities. Nucleic Acids Research 45, 11144-11158 (2017).
- Huber, F., Meurer, M., Bunina, D., Kats, I., Maeder, C.I., Stefl, M., Mongis, C., and Knop, M. PCR Duplication: A One-Step Cloning-Free Method to Generate Duplicated Chromosomal Loci and Interference-Free Expression Reporters in Yeast. PLoS ONE 9, e114590 (2014).
Credits
Special thanks go to Daria Bunina and Martin Štefl both of which are shared first authors on two and one of the main papers, respectively.